La enfermedad de Huntington (EH) fue descrita en 1872 por George Huntington, se trata de una enfermedad hereditaria poco frecuente de curso y pronóstico grave (1). Se trata de una entidad muy compleja que precisa de diversas especialidades médicas para su correcto manejo. Gracias a la colaboración de la unidad de psicosomática y psiquiatría de enlace, junto con el servicio de neurología, nace la Unidad Funcional de Enfermedad de Huntington del hospital. En ella trabaja un psiquiatra, un neurólogo y un trabajador social. Presentamos el caso de un paciente con antecedentes familiares de EH que inicia un cuadro de alteraciones conductuales por lo que es derivado a psiquiatría para valoración. Se ha obtenido el consentimiento informado para su publicación.
CASO CLÍNICO
Anamnesis: Se trata de un varón de 16 años con antecedentes familiares de EH que es derivado por neurología para valoración de alteraciones conductuales y caracteriales para descartar inicio de la enfermedad. El paciente presenta reacciones de ira puntuales y autolimitadas, así como enfrentamientos verbales con su madre en situaciones de tensión y recriminación por parte de ella. Presenta un carácter irritable con episodios de agresividad verbal y física hacia objetos. Mal rendimiento escolar en los últimos dos cursos académicos (repite 4º ESO). Este hecho ha coincidido con su traslado de residencia desde su ciudad natal a una nueva localidad y el inicio de convivencia con la pareja de su madre con quien mantiene una relación disarmónica. Refiere también tics motores simples que se presentan en estados de mayor ansiedad pero que logra controlar.
Antecedentes familiares: Padre fallecido a los 46 años (por cirrosis, se desconoce si tenía la mutación del gen para la EH), abuela paterna fallecida de EH. La abuela paterna fue madre de 7 hijos, de los cuales 5 tenían la mutación EH, de estos 5, 2 fallecieron por la enfermedad. Los otros dos hijos se desconoce si tienen la mutación, uno de ellos vive sin patología aparente, y el otro es el padre del paciente que falleció por enfermedad hepática. Prima de 8 años con inicio de EH. Hermana de 29 años con test genético negativo.
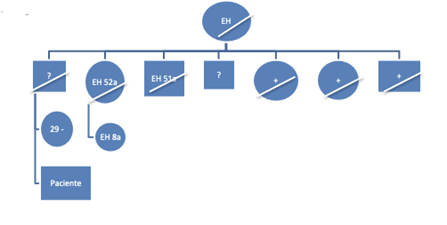
Antecedentes médicos personales: Sin antecedentes psiquiátricos personales. Sin otros antecedentes médicos destacables. Actualmente se desconoce si el paciente tiene la mutación de EH, ya que las pruebas de ADN no se realizan en menores de 18 años presintomáticos.
Exploraciones:
Exploración psicopatológica: consciente, alerta y orientado. No clínica afectiva mayor. No clínica psicótica. Sin alteraciones en la esfera instinto-vegetativa. Incremento de irritabilidad ante frustraciones a nivel familiar. Juicio de realidad conservado.
Exploración neurológica: se pasa la escala Unified Huntington’s Disease Rating Scale- Movement (UHDRS-M) con un resultado de 0, es decir no se evidencia deterioro a nivel motor. No corea, ni distonía, ni disartria, tampoco alteraciones del equilibrio ni bradicinesia. Capacidad funcional, Unified Huntington Disease Rating Scale (UHDRS-TFC): 13 (alta). No se evidencia deterioro cognoscitivo.
Juicios diagnósticos: EHJ vs alteraciones de conducta en relación con la adolescencia.
Tratamientos y curso clínico: continuar en seguimiento por consultas de neurología cada 6 meses, con 18 años podrá realizarse la prueba de test genético para determinar la mutación.
DISCUSIÓN
La EHJ es una forma de EH, caracterizada por la aparición de signos y síntomas antes de los 20 años de edad (1). Se estima una prevalencia de un 6% del total de casos de EH (2). La EH está causada por una mutación en el gen huntingtina. En el 75% de los pacientes con EHJ el padre es el progenitor afecto. Se caracteriza por la aparición de dificultades de aprendizaje, así como por hipocinesia y bradicinesia dentro del trastorno motor, siendo frecuentes las convulsiones, la ataxia y la pérdida ponderal (3). La corea, signo clásico de la EH no se observa frecuentemente en la EHJ (2). El inicio y la progresión en estos pacientes es distinto al de los adultos, suelen padecer cambios en la conducta con dificultad para prestar atención, disminución rápida y significativa del desempeño escolar, así como problemas de conducta. La progresión suele ser más rápida que en el caso de la EH en adultos (3).
El diagnóstico se realiza con clínica compatible en un individuo con un progenitor con EH probada, y se confirma por pruebas de ADN (4). Las pruebas de ADN no se realizan en pacientes menores de 18 años presintomáticos.
Las alteraciones psiquiátricas son frecuentes en los pacientes con EH, el trastorno más frecuente es la depresión en forma de irritabilidad, apatía, aislamiento social, astenia etc. Si bien, también son típicos de esta patología el trastorno obsesivo-compulsivo, la manía y el trastorno bipolar (5). Inicialmente, en la etapa presintomática puede haber cambios en el estado de ánimo y en la personalidad, con difícil diagnóstico puesto que el paciente no es consciente de estos síntomas (6).
El diagnóstico diferencial en este paciente se encontraría entre la EHJ o una alteración de conducta en un adolescente. Dada la exploración psicopatológica del paciente no existen criterios clínicos para realizar ningún diagnóstico psiquiátrico ninguno por lo que podríamos descartar un cuadro psicopatológico primario. Parece tratarse más de alteraciones relacionadas con la adolescencia y cambios de domicilio y de vinculación, sin que se aprecien síntomas compatibles en estos momentos con EHJ. Dicha sintomatología nos dirige hacia una crisis adolescente secundaria a acontecimientos estresantes. Según protocolo podría realizarse test genético con 18 años para conocer si existe mutación del gen hutingtina. Si bien es cierto, podría suscitar la duda de si en este caso estaría indicado realizar un diagnóstico genético antes de la edad adulta dados los antecedentes familiares. No existen signos ni síntomas motores que hagan sospechar enfermedad primaria neurológica y la repercusión de un test genético positivo es muy grande, teniendo en cuenta que el paciente no ha llegado a una edad de madurez emocional que le permita integrar este diagnóstico. Por lo que éticamente no creemos que estuviera justificado realizarlo.
En la unidad funcional de EH de dicho hospital fueron atendidos 41 pacientes en el año 2020. El perfil clínico de estos pacientes se caracteriza por una edad media de 49,7 años, con una proporción de 19 hombres/22 mujeres. Del total de pacientes, 3 tuvieron que ser hospitalizados (2 por infecciones respiratorias y 1 tras intento autolítico). Inicialmente todos los pacientes fueron atendidos por Neurología. El 63,4% precisaron valoración por Psiquiatría, el 39% requirió de intervención por Trabajo Social, el 21% por Endocrinología, el 21% consultó con Rehabilitación y el 9,7% por con Ginecología. Es por todo ello, que se trata de una enfermedad neurológica, psiquiátrica y genética que precisa de una adecuada coordinación de múltiples actuaciones terapéuticas, dentro de un equipo multidisciplinar en donde no sólo es necesario atender aspectos somáticos y psiquiátricos, sino también sociales y judiciales.